
Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
Polymyositis og dermatomyositis: årsager, symptomer, diagnose, behandling
Medicinsk ekspert af artiklen

Sidst revideret: 05.07.2025
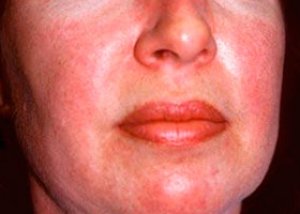
Polymyositis og dermatomyositis er sjældne systemiske reumatiske sygdomme, der er karakteriseret ved inflammatoriske og degenerative forandringer i muskler (polymyositis) eller muskler og hud (dermatomyositis). Den mest specifikke hudmanifestation er heliotrop udslæt.
Muskelpåvirkningen er symmetrisk og omfatter svaghed, en vis ømhed og efterfølgende atrofi af de proximale bækkenmuskler. Komplikationer kan omfatte visceral påvirkning og malignitet. Diagnosen er baseret på klinisk præsentation og vurdering af muskeldysfunktion ved at måle enzymniveauer, udføre MR, elektromyografi og muskelbiopsi. Behandlingen involverer glukokortikoider, undertiden i kombination med immunsuppressiva eller intravenøse immunglobuliner.
Kvinder bliver syge dobbelt så ofte som mænd. Sygdommen kan forekomme i alle aldre, men opdages oftest i alderen 40 til 60 år; hos børn - fra 5 til 15 år.
 [
[ Hvad forårsager dermatomyositis og polymyositis?
Årsagen til sygdommen menes at være en autoimmun reaktion på muskelvæv hos genetisk prædisponerede individer. Sygdommen er mere almindelig i nærvær af en belastet familiehistorie og bærere af visse HLA-antigener (DR3, DR52, DR56). Mulige udløsere er viral myositis og maligne neoplasmer. Der er rapporter om påvisning af strukturer svarende til picornavirus i muskelceller; derudover kan virus fremkalde lignende sygdomme hos dyr. Sammenhængen mellem maligne tumorer og dermatomyositis (meget sjældnere end ved polymyositis) antyder, at tumorvækst også kan være en udløsende faktor for udviklingen af sygdommen som følge af initiering af autoimmune reaktioner på almindelige antigener i tumoren og muskelvævet.
Aflejringer af IgM, IgG og den tredje komponent af komplement findes i væggene af blodkarrene i skeletmuskulaturen; dette er især karakteristisk for dermatomyositis hos børn. Patienter med polymyositis kan også udvikle andre autoimmune processer.
Patofysiologi af dermatomyositis og polymyositis
Patologiske forandringer omfatter celleskader og atrofi på baggrund af inflammation af varierende sværhedsgrad. Musklerne i øvre og nedre ekstremiteter, såvel som ansigtet, er beskadiget i mindre grad end andre skeletmuskler. Skader på de viscerale muskler i svælget og den øvre del af spiserøret, sjældnere hjertet, maven eller tarmene, kan føre til dysfunktion af ovennævnte organer. Høje koncentrationer af myoglobin på grund af rhabdomyolyse kan forårsage nyreskade. Inflammatoriske forandringer i led og lunger kan også forekomme, især hos patienter, der har antisyntetase-antistoffer.
Symptomer på dermatomyositis og polymyositis
Polymyositis kan opstå akut (især hos børn) eller subakut (oftere hos voksne). Akut virusinfektion går undertiden forud for eller er en udløsende faktor for sygdommens manifestation, hvis mest almindelige manifestationer er svaghed i de proximale muskler eller hududslæt. Smerter udtrykkes i mindre grad end svaghed. Polyarthralgi, Raynauds fænomen, dysfagi, lungesygdomme, generelle symptomer (øget kropstemperatur, nedsat kropsvægt, svaghed) kan udvikle sig. Raynauds fænomen findes ofte hos patienter med samtidige bindevævssygdomme.
Muskelsvaghed kan udvikle sig over flere uger eller måneder. For at muskelsvaghed kan vise sig klinisk, skal mindst 50 % af muskelfibrene dog være påvirket (tilstedeværelsen af muskelsvaghed indikerer således progression af myositis). Patienter kan have svært ved at hæve armene over skulderhøjde, gå op ad trapper eller rejse sig fra siddende stilling. På grund af alvorlig svaghed i bækken- og skulderbæltemusklerne kan patienter være bundet til en kørestol eller seng. Hvis nakkebøjerne påvirkes, bliver det umuligt at løfte hovedet fra puden. Påvirkning af musklerne i svælget og den øvre del af spiserøret fører til synkebesvær og opstød. Musklerne i under- og overekstremiteterne og ansigtet påvirkes normalt ikke. Der kan dog udvikles kontrakturer i lemmerne.
Hududslæt observeret ved dermatomyositis er normalt mørke i farven og erytematøse. Periorbitalt ødem i lilla farve (heliotrop udslæt) er også karakteristisk. Hududslæt kan være let hævet over hudniveauet og være glat eller skællet; lokalisering af udslæt er pande, nakke, skuldre, bryst, ryg, underarme, nedre dele af skinnebenene, øjenbryn, knæområder, mediale malleoler, dorsale overflader af interfalangeale og metakarpofalangeale led, på den laterale side (Gottrons symptom). Hyperæmi af neglens base eller periferi er mulig. Deskvamativ dermatitis, ledsaget af revner, kan udvikle sig på huden på den laterale overflade af fingrene. Primære hudlæsioner forsvinder ofte uden følgevirkninger, men kan føre til sekundære forandringer såsom mørk pigmentering, atrofi, ardannelse eller vitiligo. Subkutan forkalkning kan udvikle sig, især hos børn.
Omtrent 30 % af patienterne udvikler polyarthralgi eller polyartritis, ofte ledsaget af ødem og ledeffusion. Sværhedsgraden af ledmanifestationer er dog lille. De forekommer oftere, når der påvises antistoffer mod Jo-1 eller andre syntetaser hos patienterne.
Involvering af indre organer (undtagen svælget og den øvre del af spiserøret) er mindre almindelig ved polymyositis end ved andre reumatiske sygdomme (især SLE og systemisk sklerose). I sjældne tilfælde, især ved antisyntetasesyndrom, manifesterer sygdommen sig som interstitiel pneumonitis (i form af dyspnø og hoste). Hjerterytmeforstyrrelser og ledningsforstyrrelser kan udvikle sig, men de er normalt asymptomatiske. Gastrointestinale manifestationer er mere almindelige hos børn, der også har vaskulitis, og kan omfatte opkastning med blod, melena og tarmperforation.
Hvor gør det ondt?
Klassificering af polymyositis
Der er 5 typer polymyositis.
- Primær idiopatisk polymyositis, som kan forekomme i alle aldre, involverer ikke huden.
- Primær idiopatisk dermatomyositis ligner primær idiopatisk polymyositis, men involverer huden.
- Polymyositis og dermatomyositis forbundet med maligne neoplasmer kan forekomme hos patienter i alle aldre; deres udvikling observeres oftest hos ældre patienter, såvel som hos patienter med andre bindevævssygdomme. Udviklingen af maligne neoplasmer kan observeres både inden for 2 år før og inden for 2 år efter myositis' opståen.
- Polymyositis eller dermatomyositis i børneårene er forbundet med systemisk vaskulitis.
- Polymyositis og dermatomyositis kan også forekomme hos patienter med andre bindevævssygdomme, oftest progressiv systemisk sklerose, blandet bindevævssygdom og SLE.
Det er forkert at inkludere myositis i truncalmuskulaturen i gruppen af polymyositis, da sidstnævnte er en separat sygdom, der er karakteriseret ved kliniske manifestationer, der ligner dem ved kronisk idiopatisk polymyositis. Den udvikler sig dog i alderdommen, påvirker ofte musklerne i kroppens distale dele (f.eks. øvre og nedre ekstremiteter), har en længere varighed, reagerer mindre godt på behandling og er karakteriseret ved et typisk histologisk billede.
Diagnose af dermatomyositis og polymyositis
Polymyositis bør mistænkes hos patienter med klager over proksimal muskelsvaghed, med eller uden ømhed. Udredning for dermatomyositis er nødvendig hos patienter med klager over et udslæt, der ligner heliotrop eller Gottrons tegn, samt hos patienter med manifestationer af polymyositis kombineret med hudlæsioner, der er forenelige med dermatomyositis. De kliniske manifestationer af polymyositis og dermatomyositis kan ligne dem ved systemisk sklerose eller, mindre almindeligt, SLE eller vaskulitis. Sikkerheden af diagnosen øges ved at opfylde så mange af følgende fem kriterier som muligt:
- svaghed i de proksimale muskler;
- karakteristiske hududslæt;
- øget aktivitet af muskelvævsenzymer (kreatinkinase eller, i mangel af en stigning i dens aktivitet, aminotransferaser eller aldolase);
- karakteristiske ændringer i myografi eller MR;
- karakteristiske histologiske ændringer i en muskelvævsbiopsi (absolut kriterium).
Muskelbiopsi kan udelukke nogle klinisk lignende tilstande, såsom myositis i truncusmuskulaturen og rabdomyolyse på grund af virusinfektion. Ændringer afsløret ved histologisk undersøgelse kan variere, men kronisk inflammation, fokus på muskeldegeneration og regeneration er typiske. En nøjagtig diagnose (normalt ved histologisk verifikation) er nødvendig, før potentielt toksisk behandling påbegyndes. MR kan detektere fokus på ødem og inflammation i musklerne, efterfulgt af målrettet biopsi af disse.
Laboratorieundersøgelser kan bekræfte eller tværtimod fjerne mistanken om sygdommens tilstedeværelse og er også nyttige til at vurdere dens sværhedsgrad, muligheden for en kombination med andre lignende patologier og diagnosticere komplikationer. Selvom antinukleære antistoffer påvises hos nogle patienter, er dette fænomen mere typisk for andre bindevævssygdomme. Omkring 60% af patienterne har antistoffer mod cellekernens (PM-1) eller hele thymusceller og Jo-1. Autoantistoffers rolle i sygdommens patogenese er fortsat uklar, selvom det er kendt, at antistoffer mod Jo-1 er en specifik markør for antisyntetasesyndrom, herunder fibroserende alveolitis, lungefibrose, gigt og Raynauds fænomen.
Periodisk vurdering af kreatinkinaseaktivitet er nyttig til overvågning af behandlingen. Hos patienter med alvorlig muskelsvind kan enzymaktiviteten dog være normal på trods af tilstedeværelsen af kronisk aktiv myositis. MR-scanning, muskelbiopsi eller forhøjet kreatinkinaseaktivitet er ofte nyttige til at skelne mellem tilbagefald af polymyositis og glukokortikoid-induceret myopati.
Da mange patienter har udiagnosticerede maligniteter, anbefaler nogle forfattere screening af alle voksne med dermatomyositis og personer med polymyositis over 60 år efter følgende skema: fysisk undersøgelse, herunder bryst-, bækken- og endetarmsundersøgelser (inklusive afføringstest for okkult blod); fuldstændig blodtælling; blodkemi; mammografi; test af karcinoembryonisk antigen; urinanalyse; røntgen af thorax. Nogle forfattere sætter spørgsmålstegn ved behovet for en sådan screening hos yngre patienter, der ikke har kliniske tegn på malignitet.
Hvad skal man undersøge?
Hvordan man undersøger?
Behandling af dermatomyositis og polymyositis
Indtil inflammationen er lindret, bør fysisk aktivitet begrænses. Glukokortikoider er førstelinjelægemidler. I sygdommens akutte fase bør voksne patienter ordineres prednisolon (oralt) i en dosis på 40 til 60 mg dagligt. Regelmæssig bestemmelse af kreatinkinaseaktivitet er en tidlig indikator for effektivitet: hos de fleste patienter ses et fald eller en normalisering inden for 6 til 12 uger efter en stigning i muskelstyrke. Efter normalisering af enzymaktiviteten reduceres prednisolondosis: først med ca. 2,5 mg dagligt i en uge, derefter hurtigere; hvis den øgede muskelenzymaktivitet vender tilbage, øges hormondosis igen. Patienter, der er kommet sig, kan undvære glukokortikoider, men oftest har voksne patienter brug for langvarig glukokortikoidbehandling (10-15 mg prednisolon dagligt). Den initiale dosis af prednisolon til børn er 30-60 mg/m² én gang dagligt. Ved remission i > 1 år hos børn kan glukokortikoidbehandlingen seponeres.
I nogle tilfælde oplever patienter, der får høje doser glukokortikoider, en pludselig stigning i muskelsvaghed, hvilket kan være forbundet med udviklingen af glukokortikoidmyopati.
I tilfælde af utilstrækkelig respons på glukokortikoidbehandling, samt i tilfælde af udvikling af glukokortikoidmyopati eller andre komplikationer, der kræver dosisreduktion eller seponering af prednisolon, bør immunsuppressive midler (methotrexat, cyclophosphamid, azathioprin, cyclosporin) anvendes. Nogle patienter modtager muligvis kun methotrexat (normalt i doser, der overstiger dem, der anvendes til behandling af leddegigt) i mere end 5 år. Intravenøse immunglobuliner kan være effektive hos patienter, der er refraktære over for lægemiddelbehandling, men deres anvendelse øger behandlingsomkostningerne.
Myositis forbundet med primære og metastatiske tumorer, såvel som myositis i truncusmusklerne, er normalt mere refraktære over for glukokortikoidbehandling. Remission af myositis forbundet med maligne tumorer er mulig efter fjernelse af tumoren.
Hvad er prognosen for dermatomyositis og polymyositis?
Langvarig remission (og endda klinisk bedring) over 5 år observeres hos mere end halvdelen af de behandlede patienter; hos børn er dette tal højere. Tilbagefald kan dog forekomme når som helst. Den samlede femårsoverlevelsesrate er 75 %, højere hos børn. Dødsårsagerne hos voksne er alvorlig og progressiv muskelsvaghed, dysfagi, nedsat ernæring, aspirationspneumoni eller respirationssvigt på grund af lungeinfektioner. Polymyositis er mere alvorlig og resistent over for behandling, hvis der er skade på hjerte og lunger. Død hos børn kan forekomme på grund af intestinal vaskulitis. Den generelle prognose for sygdommen bestemmes også af tilstedeværelsen af maligne neoplasmer.