
Alt iLive-indhold gennemgås medie eller kontrolleres for at sikre så meget faktuel nøjagtighed som muligt.
Vi har strenge sourcing retningslinjer og kun link til velrenommerede medie websteder, akademiske forskningsinstitutioner og, når det er muligt, medicinsk peer reviewed undersøgelser. Bemærk at tallene inden for parentes ([1], [2] osv.) Er klikbare links til disse undersøgelser.
Hvis du mener, at noget af vores indhold er unøjagtigt, forældet eller på anden måde tvivlsomt, skal du vælge det og trykke på Ctrl + Enter.
Aper-syndromet
Medicinsk ekspert af artiklen

Sidst revideret: 04.07.2025

 [
[ Årsager Aper-syndromet
Udviklingen af syndromet fremkaldes af en forstyrrelse i strukturen af et gen placeret på kromosom 10. Dette gen er ansvarligt for processen med fingerseparation, og derudover for rettidig lukning af kraniale suturer.
Derudover anses infektionssygdomme, der opstår under graviditeten (såsom røde hunde eller tuberkulose, syfilis eller influenza samt meningitis), og moderens eksponering for røntgenstråler for at være årsagerne til patologien. Oftest opdages et sådant syndrom hos børn født af ældre forældre.
Patogenese
Apert syndrom er arveligt. Dets type er autosomal dominant (dvs. i en familie, hvor en af de kommende forældre er syg, er sandsynligheden for at få et barn med denne sygdom 50-100%).
En unik mutation i fibroblastvækstfaktorreceptor 2 (FGFR2) resulterer i et øget antal progenitorceller, der udvikles langs den osteogene bane. Dette resulterer i sidste ende i øget dannelse af subperiosteal knoglematrix og for tidlig ossifikation af kraniesuturerne under fosterudviklingen. Rækkefølgen og hastigheden af sutursmeltningen bestemmer graden af deformitet og invaliditet. Når suturmaterialet er helet, er væksten af andet væv vinkelret på denne sutur begrænset, og de sammenvoksede knogler fungerer som et enkelt knoglestillads.
Det første genetiske bevis for, at syndaktyli ved Apert syndrom skyldes en defekt i keratinocytvækstfaktorreceptoren (KGFR), var observationen af en korrelation mellem KGFR-ekspression i fibroblaster og sværhedsgraden af syndaktyli.
Amblyopi og strabismus er mere almindelige hos patienter med FGFR2 Ser252Trp-mutationen, og diskatrofi er mere almindelig hos patienter med FGFR2 Pro253Arg-mutationen. Patienter med FGR2 Ser252Trp-mutationer har en signifikant højere prævalens af synshandicap sammenlignet med patienter med FGFR2 Pro253Arg-mutationen.
Symptomer Aper-syndromet
Nogle manifestationer af sygdommen er tydeligt synlige allerede ved barnets fødsel, fordi de udvikler sig inde i moderens livmoder. Blandt de vigtigste symptomer på syndromet:
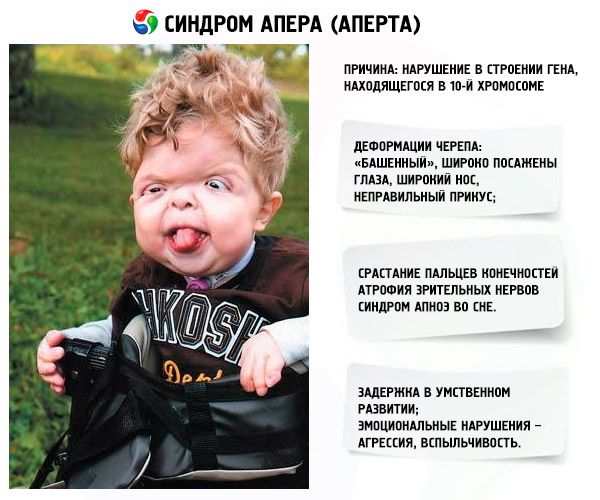
- Kraniet er deformeret - det strækker sig i højden og får et "tårn"-udseende; derudover er øjnene bredt ansat og let udstående (fordi øjenhulernes størrelse mindskes); næsen udvider sig, og der dannes et forkert bid (de øvre tænder stikker for meget ud);
- Fingrene på lemmerne er fuldstændig sammenvoksede (primært ringfingeren, såvel som langfingeren og pegefingeren) og ligner en hudmembran eller en fuldstændig sammenvoksning af knogler; derudover kan der vokse ekstra fingre;
- Mental retardering (forekommer ikke hos alle);
- Atrofi af synsnerven, hvilket resulterer i et fald i synsskarpheden (i nogle tilfælde udvikles fuldstændigt synstab);
- Øget intrakranielt tryk, som opstår som følge af for tidlig lukning af kraniale suturer, manifesterer sig i form af hovedpine og opkastning med kvalme;
- Da overkæben forbliver underudviklet, opstår der vejrtrækningsproblemer;
- Søvnapnøsyndrom er en almindelig forekomst.
- Følelsesmæssige manifestationer – aggression, mangel på selvkontrol, stærk irritabilitet.
Komplikationer og konsekvenser
De fleste patienter har en eller anden grad af obstruktion af de øvre luftveje i spædbarnsalderen. De øvre luftveje er dårligt patenterede på grund af den reducerede størrelse af nasopharynx og choanae, og de nedre luftveje kan forårsage tidlig død på grund af abnormiteter i luftrørsbrusken.
Diagnosticering Aper-syndromet
For at stille en diagnose kræves følgende trin:
- Lægen bør analysere patientens klager samt sygehistorien. Det er nødvendigt at finde ud af, om der har været tilfælde af lignende patologi i familien;
- Neurologisk undersøgelse for at vurdere kraniets form og patientens intellektuelle udvikling (særlige spørgeskemaer samt interview);
- Undersøgelse af fundus for at detektere tilstedeværelsen af symptomer på øget intrakranielt tryk (hævelse af den optiske disk samt sløring af dens kanter);
- For at vurdere kraniets tilstand udføres et røntgenbillede;
- Computer- og magnetisk resonansbilleddannelse af hovedet for at undersøge hjernens og kraniets struktur lag for lag, for at bestemme tilstedeværelsen af symptomer på for tidlig fusion af kraniale suturer, og derudover hydrocephalus (på grund af øget intrakranielt tryk akkumuleres overskydende cerebrospinalvæske (dette er cerebrospinalvæsken, der fremmer metabolisk proces, såvel som ernæring af hjernen));
- Røntgenbillede af fødder og hænder for at bestemme årsagen til fingrenes sammenvoksning (dette er vigtigt for planlægning af efterfølgende kirurgisk indgreb);
- En konsultation med en medicinsk genetiker og en neurokirurg kan ordineres.
Test
Der udføres genetisk analyse af almindelige mutationer, der forekommer i FGFR2-typegenet.
Differential diagnose
Dette syndrom skal differentieres fra andre genetiske patologier, hvor der observeres kraniosynostose. Det drejer sig om sygdomme som Pfeiffer-, Crouzon-, Saethre-Chotzen- og Carpenter-syndromer. Molekylærgenetiske testmetoder anvendes til at udelukke disse anomalier.
Hvem skal kontakte?
Behandling Aper-syndromet
Kirurgi betragtes som den eneste effektive behandling for Apert syndrom – det hjælper med at korrigere individuelle fysiske defekter og korrigerer også mental retardering.
Under denne procedure lukkes den koronale sutur for at forhindre mulig hjerneskade. Den mest almindelige teknik er kraniofacial distraktion, som involverer gradvis kranietraktion. Ortodontisk og/eller ortognatisk kirurgi udføres for at fjerne individuelle ansigtsdefekter.
Derudover gennemgår patienterne kirurgisk korrektion af fingerfusionen.
Referencer
Medicinsk genetik. National guide. Redigeret af Ginter EK, Puzyrev VP GEOTAR-Media. 2022